Usage¶
COMET can be executed with a single terminal command:
$ hgmd [-h] [-X [X]] [-L [L]] input_path output_path
- The
-hflag prints a help message, explaining each argument. - The
-Xand-Lflags accept argumentsXandL. These are the bounds used in calculation of the gene expression cutoff value. TheXargument specifies the minimum number of expressing cells that should be in the cluster; the cutoff value is selected such that there are at minimumXcells both in the cluster and expressing any gene of interest. Similar, theLargument is the maximum number of expressing cells across the entire population; the cutoff value is selected such that no greater thanLcells express any gene of interest. These flags may be omitted, or used one at a time. They default to having no bound. - The
input_pathargument specifies the directory where input files are found. See below for instructions on formatting these files. - The
output_pathargument specifies the directory where COMET will put its output files.
For examples of COMET usage and output, see Examples.
Input formatting¶
Before you use COMET with gene expression data, your data should be formatted into 3 files:
markers.txt: a table in CSV format. The first row of the table lists gene names, while the first column lists cell names (the cell at the very top left should be blank). Each element in the rest of the table should contain a numerical gene expression value, corresponding to the row/cell and column/gene of the element.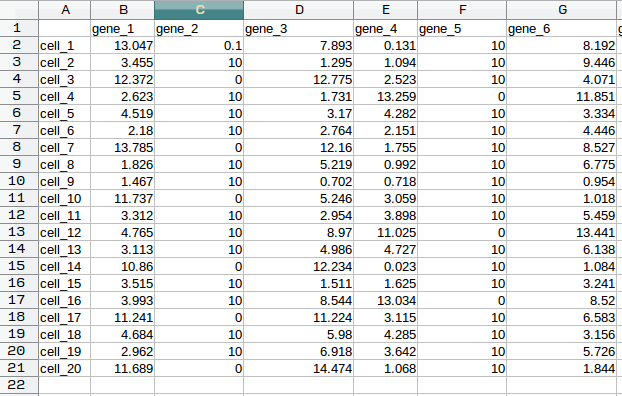
tsne.txt: a table in CSV format, of three columns without column labels. The first column is cell name (the same as those inmarkers.txt), the second is the tSNE_1 value for the cell, and the third is the tSNE_2 value for the cell.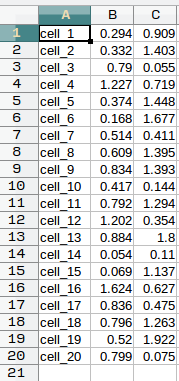
cluster.txt: a table in CSV format, of two columns without column labels. The first column is cell name (consistent withmarkers.txtandtsne.txt), and the second is the cluster of which the cell is a member.